Newsletter
Kinetic analysis of covalent and irreversible inhibitors

Carna Biosciences Newsletter Vol. 8
Covalent inhibitors generally contain electrophiles that interact covalently with nucleophilic residues such as serine, lysine, histidine and cysteine in enzymes (1). Penicillin, discovered in 1928 as the first effective treatment for bacterial infections, is one of the best known examples of a covalent inhibitor. The β-lactam present in penicillin binds covalently to the active site serine of the bacterial enzyme DD-transpeptidase and thus prevents the bacteria from synthesizing their essential cell-wall (2). The development of covalent inhibitors has advanced significantly in recent years, and now approximately 30% of marketed drugs are covalent inhibitors.
Advantages and disadvantages of covalent inhibitors
Compared to reversible inhibitors, covalent inhibitors provide advantages such as strong target affinity (3) and long-term therapeutic effect. Despite these benefits, however, there can be serious issues resulting from their use in that a covalent inhibitor can induce significant toxicity when it interacts with nucleophilic residues in off-target proteins.
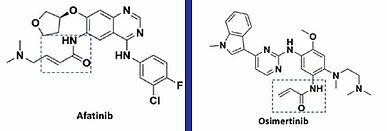
Most covalent inhibitors that have been used for many years, like aspirin or penicillin, were fortuitously discovered by phenotypic screening without elucidating their mechanism of action. More recently, it has become possible to design covalent inhibitors that selectively bind to the target protein, allowing greater focus on development of small molecules that can suppress non-specific interactions. Representative examples of effective covalent kinase inhibitors in this category are the EGFR inhibitors afatinib and osimertinib, used for the treatment of non-small cell lung cancer (NSCLC). Both contain acrylamide (Michael acceptor: α, β-unsaturated carbonyl group), which forms a covalent bond with Cys797 located in the EGFR active site, thus irreversibly inhibiting EGFR (4) (Fig.1). Afatinib was approved by the FDA in 2013 as the first covalent kinase inhibitor. Osimertinib prolonged patients’ progression-free survival (PFS) compared to the 1st and 2nd generation EGFR inhibitors and effectively inhibited the drug resistant mutant EGFR [T790M]. As a result, osimertinib was approved by the FDA in 2015.
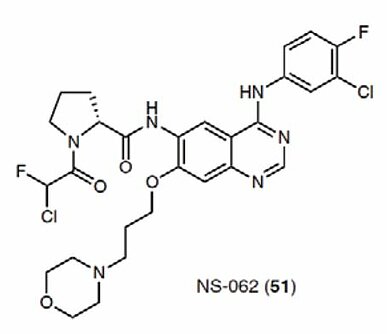
Most targeted covalent protein kinase inhibitors approved to date also employ acrylamide as a warhead, which forms a covalent bond with cysteines. However, it has been reported that certain types of acrylamide-based kinase inhibitors interact with off-target proteins (5). Ojida et al. at Kyushu University found α-chlorofluoroacetamide (CFA) as a new electrophilic reactive group that modifies Cys residues. In the work, various CFA-appended quinazoline derivatives based on the quinazoline contained in afatinib were synthesized, and the findings indicated that the derivative interacts with EGFR more selectively than the corresponding Michael acceptors (6). This was attributed to the covalent bond between CFA and Cys, which is susceptible to hydrolysis under neutral aqueous conditions but is stable in the solvent-inaccessible ATP-binding pocket of EGFR. NS-062, a CFA-modified quinazoline, effectively suppressed tumor growth in a mouse xenograft model similarly to afatinib, however it did not lead to the loss of body weight observed with afatinib. CFA is a promising agent that may be incorporated into novel small molecules that are eventually advanced to the clinic for the treatment of diseases.
Quantifaction of inhibition potential
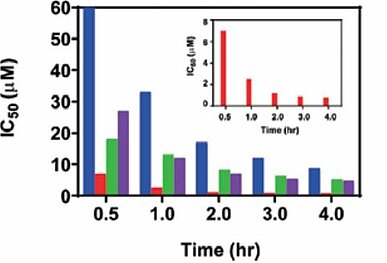
The potency of enzyme inhibition by a drug is frequently quantified in terms of IC50 values. This is suitable for reversible inhibitors where the IC50 value is a measure of binding affinity under equilibrium conditions. However, the IC50 value is often an unsuitable approach to determine inhibitory activity of irreversible covalent inhibitors. Given that irreversible inhibitors permanently bind to the target enzyme, the functional catalytic enzyme in the system decreases as time advances. As a result, the IC50 value of an irreversible inhibitor decreases time-dependently (7)8)(Fig.3). In addition, since the lowest limit of IC50 value is half the enzyme concentration, there is a risk that the true potency may not be evaluated correctly for a very potent inhibitor with enzyme inhibitory activity greater than the system can quantify. Therefore, additional approaches are required to accurately assess the potency of covalent inhibitors.
Kinetic assays have attracted attention as a means to quantify the activity of covalent inhibitors. Kinetically speaking, the binding of most irreversible inhibitors can be described by the mechanism shown in Scheme 1. Here, the reversible binding of inhibitor to enzyme is characterized by Ki in the first step, and an irreversible, covalent modification is described by the maximum potential rate, kinact, in the second step. The ratio of kinact/Ki has the form of a second-order rate constant and can be used to compare the inactivation kinetics of different irreversible, covalent inhibitors. Gilead Sciences has used the kinetic measurement technology of our collaborator, AssayQuant Technologies, to obtain kinact/Ki values. The kinact/Ki values and IC50 values were compared for the purpose of assessing their irreversible BTK inhibitor, tirabrutinib, for selectivity relative to other irreversible BTK inhibitors. The investigators reported that selectivity assessment using IC5 0 values tended to exaggerate selectivity relative to the selectivity measured by kinact/Ki8), demonstrating the value of this approach.
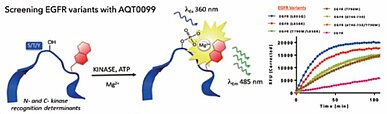
Many of AssayQuant Technology’s kinetics assays using PhosphoSens™ technology, such as those presented in this newsletter, have been developed using Carna’s kinase products (Fig.4). Carna also offers inhibitor residence time determinations for many kinases using our NanoBRET™ (TE) Intracellular Kinase Cell-Based Assay Services as a tool to explore the mode of inhibition of known drugs and potential drug candidates. Please contact us if you are interested in learning more about our kinase products or cell-based binding assay services.
References:
- Eur J Med Chem. 2017; 138:96-114. De Cesco S.
- RSC Med Chem. 2020; 11:876-884. Sutanto F.
- J Med Chem. 2009; 52(2):225-33. Smith AJ.
- ChemMedChem. 2019; 14(9):889-906. Ghosh AK.
- Nat Chem Biol. 2014; 10(9):760-767. Lanning BR.
- Nat Chem Biol. 2019; 15(3):250-258. Shindo N., Ojida A.
- J Biomol Screen. 2009; 14(8):913-23. Krippendorff BF.
- Biochim Biophys Acta Gen Subj. 2020; 1864(4):129531. Liclican A.
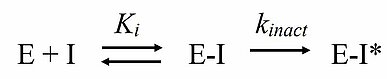
Further news
-
Newsletter
Cancer treatments using inhibitors of CDK, a cell cycle regulator
Read moreCarna Newsletter Vol.16
-
Newsletter
ALK drug resistant mutations: Challenges for the treatment of lung cancer
Read moreCarna Newsletter Vol.15
-
Technical Note
DGKα and DGKζ are key targets for cancer immunotherapy
Read moreCarna Newsletter Vol.14